





Insulin resistance is among the most prevalent endocrine derangements in the world, and it is closely associated with major diseases of global reach including diabetes mellitus, atherosclerosis, nonalcoholic fatty liver disease, and ovulatory dysfunction. It is most commonly found in those with obesity but may also occur in an unusually severe form in rare patients with monogenic defects. Such patients may loosely be grouped into those with primary disorders of insulin signaling and those with defects in adipose tissue development or function (lipodystrophy). The severe insulin resistance of both subgroups puts patients at risk of accelerated complications and poses severe challenges in clinical management. However, the clinical disorders produced by different genetic defects are often biochemically and clinically distinct and are associated with distinct risks of complications. This means that optimal management of affected patients should take into account the specific natural history of each condition. In clinical practice, they are often underdiagnosed, however, with low rates of identification of the underlying genetic defect, a problem compounded by confusing and overlapping nomenclature and classification. We now review recent developments in understanding of genetic forms of severe insulin resistance and/or lipodystrophy and suggest a revised classification based on growing knowledge of the underlying pathophysiology.
Introduction
Definition and Prevalence of Severe IR
Generic Clinical Features of Severe IR
Abnormal glucose homeostasis
Ovarian dysfunction
Acanthosis nigricans
Clinical Features Limited to Some Severe IR Subtypes
Dyslipidemia and hepatic steatosis
Abnormal adipose development or topography
Growth disorders
Sexual Dimorphism in Severe IR
Biochemical Subphenotyping of Severe IR
Monogenic IR Classification/Nomenclature
The INSR Spectrum
Downstream Insulin Signaling Defects
Disorders of Adipose Tissue Development/Function (Lipodystrophies)
Digenic IR
Complex Syndromes
Therapy
Dietary and lifestyle modification
Insulin sensitization and replacement
Adipose tissue offloading
Summary
I Introduction
Insulin resistance (IR), or more precisely the reduced responsiveness of the body to the glucose-lowering activity of insulin, is closely associated with some of the most prevalent chronic clinical disorders, namely type 2 diabetes, atherosclerosis, polycystic ovarian syndrome, and hepatic steatosis. The population-wide toll of morbidity and mortality attributable to IR is large and growing, in particular due to the consequences of coronary artery disease (1), type 2 diabetes (2), polycystic ovary syndrome (3), and fatty liver disease, and indeed IR is a cardinal feature of the metabolic syndrome itself (4).
Although IR is a trait with significant heritability (5–8), it is usually only clinically expressed in the context of obesity, especially where this has a predominantly centripetal distribution. In a small number of patients, however, IR of an unusually severe degree develops without obesity, or in association with generalized or regional lack of adipose tissue. Many such patients harbor pathogenic single gene mutations, and several of these have been identified in recent years. These genetic defects may currently be grouped into those affecting insulin signaling and those affecting adipocyte development and/or function. Detailed physiological study of such patients with defined genetic defects has begun to identify distinct subphenotypes of IR and has yielded insights into the mechanistic basis of more prevalent forms of IR.
Severe IR may also arise through acquired, immune-mediated mechanisms. These include antibodies against either insulin or the insulin receptor leading to blockade of insulin action (9) or autoimmune destruction of adipocytes leading to lipodystrophy (10, 11). Many, but not all, of these patients have coexisting autoimmune disease, alerting physicians to the possibility of acquired severe IR. These disorders have been recently reviewed (12–15).
The prevailing clinical nomenclature in the field of severe IR dates from early work on these syndromes in the 1970s (9, 16). We now describe the current state of knowledge of genetic forms of severe IR, suggest a refined classification based on recent findings, and review currently available treatments.
II Definition and Prevalence of Severe IR
Plasma insulin, whether determined in the fasting state or after a glucose challenge, is a continuous variable, and so thresholds used to diagnose IR and “severe” IR are arbitrary. Furthermore, such thresholds are only reliable before β-cell decompensation has occurred. In entirely insulin-deficient individuals, severe IR may be defined solely in terms of the body mass-adjusted requirements for exogenous insulin to maintain euglycemia, whereas in nondiabetic patients with compensated IR, severe IR may be defined solely in terms of plasma insulin levels before and/or after a glucose challenge, with reference to data from a control population. However, between these extremes, in patients with relative rather than absolute insulin deficiency, diagnosis of severe IR is based on semiquantitative assessment of the biochemical abnormality coupled with clinical evidence of severe IR. A further complication is that severe IR is most commonly seen in obese patients, and yet they are a group far less likely to harbor single gene defects. Similarly, puberty is a time of “physiological” IR, and so ideally biochemical assessment of possible severe IR should be made with reference to normative data derived from people of similar adiposity and developmental stage. Subject to these caveats, a suggested working diagnostic scheme for likely monogenic severe IR is shown in Fig. 1. This emphasizes that, whereas numerical determinants of severe IR have utility in the settings of normal glycemia and absolute insulin deficiency, diagnosis in the context of partial β-cell decompensation, which is the most common scenario, relies heavily on interpretation of physical signs and clinical history. Although not widely employed in current clinical practice, we have also found a nomogram derived from a large, nondiabetic population, showing the relationship between body mass index and insulin levels to be helpful in discriminating degrees of IR in overweight/obese patients that are manifestly disproportionate to the degree of adiposity and are thus more likely to have a contribution from a single gene defect (Fig. 1B).
![Diagnosis of possible monogenic severe IR. A, Suggested (arbitrary) diagnostic criteria. B, Relationship between body mass index (B.M.I.) and fasting plasma insulin in a healthy European nondiabetic population (n = 800). The solid line represents the 50th centile, and the dashed lines the 5th and 95th centiles. [Figure courtesy of Prof. Nicholas J. Wareham.].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/edrv/32/4/10.1210_er.2010-0020/3/m_zef0041127830001.jpeg?Expires=1716349448&Signature=oPmPNYN8-jeucPRM7hIWcuUqevCGRcTEvIwVsNU8AzNyWTdrc82P6KTLtJ~desVnqImKkWU1d~ZHaxrDIHiZGQLAorcKSdsB~U5gGMZlhIX8gczBUz~HRmIQTOjQx-nkkQJarlLz3qyAz3H3WpCOxBEtV4lG0lJvZ655NmBNY4Xvab9rVf1wYf-GPUpoh7~EN9bGKMyNeLUX2aGt63Yt9ZGWJb2wDamavUe1CZoMy8EyPSlNUzm3y59mAg~groI8EtyB2LIKAsVM8AekM0RX2yOLE8z5yqutluG6qh~f6K7q7DbGQ0B6O0hTIHX3mAEOs4HPkGDvqoiw-Pk4070uqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Diagnosis of possible monogenic severe IR. A, Suggested (arbitrary) diagnostic criteria. B, Relationship between body mass index (B.M.I.) and fasting plasma insulin in a healthy European nondiabetic population (n = 800). The solid line represents the 50th centile, and the dashed lines the 5th and 95th centiles. [Figure courtesy of Prof. Nicholas J. Wareham.].
These diagnostic complexities mean that severe IR is often not recognized, especially in men. Furthermore, patients present to many different clinical services according to their dominant clinical problem, and for these reasons no population-based prevalence figures exist. However, clinical experience suggests that approximately 0.1–0.5 of patients in hospital-based diabetes practices may have monogenic forms of severe IR.
III Generic Clinical Features of Severe IR
A Abnormal glucose homeostasis
Clinical awareness of “IR” is greatest among those caring for patients with established diabetes mellitus, where it is recognized most commonly by a requirement for large doses of exogenous insulin. However, hyperglycemia is not usually the earliest clinical manifestation of severe IR. It may be recognized much earlier by the presence of acanthosis nigricans and/or ovarian hyperandrogenism in women. Furthermore, symptomatic hypoglycemia often precedes hyperglycemia, sometimes by many years. Characteristically, hypoglycemia related to severe IR occurs postprandially, with autonomic symptoms sometimes progressing to neuroglycopenia and seizures if not abrogated by oral carbohydrate. Such severe postprandial hypoglycemia may be seen in patients with insulin receptor defects (17), insulin signal transduction defects, or primary lipodystrophies (18, 19). Its mechanism is unclear, but it most likely relates to severe impairment of hepatic insulin clearance due primarily to a insulin receptor defect or secondarily to the consequences of hepatic steatosis (20).
Even in the context of severe loss of insulin receptor function, hyperplasia of pancreatic β-cells, with attendant extreme hyperinsulinemia, may prevent hyperglycemia for many years, indicating that the receptor on islets themselves is not a prerequisite for β-cell expansion, as suggested by some murine studies (21). However, in most cases pancreatic β-cell hyperplasia does eventually fail to compensate for severe IR, and hyperglycemia ensues. Commonly, hyperglycemia diagnostic of diabetes is only seen after an oral glucose challenge, contrasting with fasting hypoglycemia or normoglycemia, making fasting glucose alone an inadequate diagnostic test for diabetes in the context of severe IR. Compounding this, glycosylated hemoglobin may be normal, or even low, at the time when postload glucose levels are diagnostic of diabetes. Although these observations suggest that the various current diagnostic criteria for diabetes may have different utilities in predicting risk of diabetic complications in severe IR, this has not yet been studied. The time taken to β-cell decompensation varies substantially, with diabetes developing in the neonatal period in the most severe cases and in the fourth decade or beyond at the milder end of the spectrum, especially in men.
B Ovarian dysfunction
Severe IR most commonly presents to clinical attention first as oligomenorrhea and severe hyperandrogenism in young women after menarche, although the underlying hyperinsulinemia is often not recognized. Ovarian ultrasonography usually reveals multiple peripheral cysts as seen in idiopathic polycystic ovary syndrome. In severe cases, cysts may become very large and vulnerable to hemorrhage or torsion, and surgical removal may be required, sometimes in infancy (Fig. 2). Hyperandrogenism in IR may be severe, with testosterone levels above 10 nmol/liter sometimes seen, well in excess of thresholds commonly reported to discriminate virilizing tumors from nontumoral hyperandrogenism (22).
![Ovarian appearances in severe IR. A, Ultrasound appearance of an ovary in a 14-yr-old patient with severe IR due to a heterozygous mutation in the insulin receptor. B, Perioperative appearance of large ovarian and Fallopian tube cysts in a patient with digenic severe IR due to heterozygous mutations in the PPARG and PPP1R3A genes (121). C and D, Ovarian histology of the same patient, showing prominent sclerosis of the superficial ovarian cortex associated with multiple follicle cysts (C) and stromal hyperthecosis with nests of eosinophilic luteinized cells (arrows) (D) embedded in hyalinized ovarian stroma. [Histological images courtesy of Dr. Merche Jimenez-Linan.].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/edrv/32/4/10.1210_er.2010-0020/3/m_zef0041127830002.jpeg?Expires=1716349448&Signature=q8dfe6JhyXhSqrSLh9SVzL0NUws15e-bEwIcypBlnAfvRLdONCdXfvpovAp-e0jhhBnAXfPSNkFIWQESPnQkjyEBk4UzJG6Ef6SvVwES0b5yBh5Ny1pFSdOiFSfDd~Ctwh~g6LMEsc-1vVLxw1a~2K~uXsqt2OXJLlMYopCd7RHOiU1v~rCRD1tVnqdMpvWs-OOimgAiV2K1XRXDYLleNpb4UGYN3jU4YLotLxFcyR3JZLfvzwBfci52~B1qlpfrxDK8PONkcx36zUk30QrK8nscwGEkAdfAcJHJjJnXAdKTRPmlrNpLod4iNwqkdKs-CjsrdEV9UhiedGnjlkds7g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Ovarian appearances in severe IR. A, Ultrasound appearance of an ovary in a 14-yr-old patient with severe IR due to a heterozygous mutation in the insulin receptor. B, Perioperative appearance of large ovarian and Fallopian tube cysts in a patient with digenic severe IR due to heterozygous mutations in the PPARG and PPP1R3A genes (121). C and D, Ovarian histology of the same patient, showing prominent sclerosis of the superficial ovarian cortex associated with multiple follicle cysts (C) and stromal hyperthecosis with nests of eosinophilic luteinized cells (arrows) (D) embedded in hyalinized ovarian stroma. [Histological images courtesy of Dr. Merche Jimenez-Linan.].
The ovarian hyperandrogenism of IR is driven by synergy between gonadotropin and insulin action on the ovary (23, 24). Thus, it may be clinically apparent during both infancy and postpubertal life, when the hypothalamic-pituitary-gonadal axis is fully active, and it may also accelerate puberty (25). Severe ovarian hyperandrogenism may occur postmenopausally, in which case ovarian histology characteristically reveals hyperthecosis, or hyperplasia of the androgen-secreting theca cells (26). However, hyperandrogenism is not seen, despite extreme hyperinsulinemia, when insulin receptor function is lost for the first time postmenopausally (12), suggesting that hyperinsulinemia around the time of the menopause may be necessary to sustain androgen-secreting theca cells.
The main differential diagnosis of IR-related severe hyperandrogenism is congenital adrenal hyperplasia or an androgen-secreting tumor, but in these cases acanthosis nigricans is not usually prominent unless the patient is also overweight or obese. Although congenital adrenal hyperplasia is easily diagnosed biochemically, discriminating autonomous androgen secretion and hence virilizing tumors may be more challenging (27). A complicating consideration is that ovarian tumors may arise in the context of sustained severe hyperinsulinemia, most likely due to chronic activation of IGF-I receptor-mediated signaling (28).
C Acanthosis nigricans
Another feature of nearly all known forms of severe IR is acanthosis nigricans, a velvety thickening of the skin. It is usually found in the axillae, nape of the neck, and groin, but it can occur in any flexures and in the most extreme cases may be periocular, perioral, perianal, or even occur on planar surfaces (Fig. 3). It is commonly associated with acrochordons (skin tags). Histologically, acanthosis nigricans is characterized by hyperkeratosis, sometimes with hyperpigmentation, as well as mild papillomatosis, suggesting that both keratinocytes and dermal fibroblasts are affected (29). The precise pathogenesis is unclear, but it may also rarely be found in congenital syndromes without IR (29) or as a paraneoplastic syndrome (30), and several lines of evidence suggest that enhanced signaling through mitogenic tyrosine kinase-type receptors including the IGF-I receptor plays a central role (29, 31). In IR, acanthosis depends on hyperinsulinemia, and this is not seen in the rare situation of “pre receptor” IR due to unusually high levels of anti-insulin antibodies in those receiving exogenous insulin therapy (32). Fading of acanthosis indicates a reduction in insulin levels either due to lessening of IR or, conversely, worsening of β-cell failure. Acanthosis nigricans may become excoriated and/or infected, and in concert with IR-related hyperandrogenism may contribute to hydradenitis suppurativa (33).
![Acanthosis nigricans (AN) in severe IR. A, Severe AN on the neck in a prepubertal patient with autosomal dominant IR of unknown cause. B, AN associated with exuberant axillary acrochordons in a 50-yr-old male with severe IR of unknown cause. C–F, AN in abdominal skin flexures of a 15-yr-old boy with severe IR due to a heterozygous INSR mutation (C), on the foot of a patient with congenital generalized lipodystrophy and severe IR due to homozygous AGPAT2 mutations (D), on the knuckles in a prepubertal patient with severe IR of unknown cause (E), and on the neck of a prepubertal girl with RMS due to a homozygous INSR mutation (F). G, Histological appearances from a nuchal skin biopsy showing characteristic papillomatosis (solid arrows), hyperkeratosis, and some acanthosis (open arrow). [Histological image courtesy of Dr. Ed Rytina.].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/edrv/32/4/10.1210_er.2010-0020/3/m_zef0041127830003.jpeg?Expires=1716349448&Signature=mJLCuAnksIYYwXCUpl8lwSm192pjoKPhaVcGpuJZ~rmDqfdBTj-KbGh3rjh0hiVLf66XU2R8xtONNfHYXKMAaTuXwqBctskM4pVzrCKz9jM5xPI42q5WlRxheHkSk2i0Y5lVyIZxqRXNIbvpwwgC0l~KL4BqH2bh64SW6L5jPcIRJgWzPfdT-5JQ7H67w0Tx~wgDH0etb1qnaH3vRO0LCapWW~SgRPYblN-0D59ex78zXhNsMJH4~zRjHEv7mM7e4Z~3HAjWWOp4tsnz-iGKc3uvbhAG4GJSuBzXXk7yBjLEbPQwT84wFp7lFh6rlFSzr43FGvzIrCr67VrqU~XuXA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Acanthosis nigricans (AN) in severe IR. A, Severe AN on the neck in a prepubertal patient with autosomal dominant IR of unknown cause. B, AN associated with exuberant axillary acrochordons in a 50-yr-old male with severe IR of unknown cause. C–F, AN in abdominal skin flexures of a 15-yr-old boy with severe IR due to a heterozygous INSR mutation (C), on the foot of a patient with congenital generalized lipodystrophy and severe IR due to homozygous AGPAT2 mutations (D), on the knuckles in a prepubertal patient with severe IR of unknown cause (E), and on the neck of a prepubertal girl with RMS due to a homozygous INSR mutation (F). G, Histological appearances from a nuchal skin biopsy showing characteristic papillomatosis (solid arrows), hyperkeratosis, and some acanthosis (open arrow). [Histological image courtesy of Dr. Ed Rytina.].
IV Clinical Features Limited to Some SevereIR Subtypes
All the above are seen in severe IR irrespective of underlying etiology, whether congenital or acquired, and whether due to a primary signaling defect or due to lipodystrophy. Some features of severe IR syndromes, in contrast, are seen in only some subtypes.
A Dyslipidemia and hepatic steatosis
Hypertriglyceridemia and low high-density lipoprotein cholesterol levels (hereafter designated “metabolic dyslipidemia”) are closely associated with prevalent forms of IR (34–37) and are seen in more severe form also in patients with severe monogenic IR. Indeed, in some cases, hypertriglyceridemia may be complicated by pancreatitis and eruptive xanthomata, and hepatic steatosis may progress to steatohepatitis, cirrhosis, and hepatocellular carcinoma (38). The presence of significant dyslipidemia and hepatic steatosis is a sensitive but nonspecific clinical indicator of underlying lipodystrophy. Their absence in a patient with severe IR is suggestive of a primary insulin receptoropathy (39, 40). Recently published mouse data (41–43), supported by our own observations in patients with severe IR (39), suggest that hepatic steatosis and dyslipidemia are a consequence of selective postreceptor (or partial-) hepatic IR (44).
B Abnormal adipose development or topography
Lipodystrophy is a common cause of severe IR and should be considered in all cases. The fact that fat mass in lean women is close to double that in lean men, coupled with the readily recognizable femorogluteal depot usually present in women, generally means that lipodystrophy is more readily detected in women than men. Lean athletic men can be very difficult to distinguish from lipodystrophic men, particularly those with partial lipodystrophy.
C Growth disorders
Severe IR per se may be associated with a range of growth disorders, including linear growth impairment (40, 45), prepubertal linear growth acceleration (46), or “pseudoacromegalic” soft tissue overgrowth in adulthood (47–50). Although the precise molecular basis of this close association between severe IR and growth abnormalities has yet to be determined, it may well relate to perturbation of the cross talk between the endocrine axis controlling growth and insulin action, or perhaps to abnormal paracrine action of IGF-I or IGF-II (45). Both IGF-I, the major hormone driving linear growth, and IGF-II, which is an important determinant of growth in utero, but which remains present at high levels in serum in postnatal life in humans but not rodents, exert mitogenic effects through the IGF-I receptor and also have significant ability to stimulate the insulin receptor. Insulin, conversely, is able to stimulate the IGF-I receptor at concentrations seen in extreme hyperinsulinemia (51, 52). Added to this direct cross talk, insulin or IR has been shown in a variety of contexts to influence action of IGF through alterations in expression of the ligands themselves, of their binding proteins, or of their receptors (53). These observations have yet to be synthesized into a coherent model accounting for growth abnormalities in severe IR; however, improving understanding of these phenomena will not only be of relevance to these rare conditions, but may also give mechanistic insights into the link between common obesity/IR and both the prevalence and prognosis of a variety of different cancers (52, 54, 55).
V Sexual Dimorphism in Severe IR
A prominent feature of severe IR is the earlier presentation and more severe metabolic derangement seen in affected women (56). A major contributor to this is hyperinsulinemia-driven ovarian dysfunction, with hyperandrogenism and oligomenorrhea serving as clinical red flags that lead to early presentation (57). Men, in contrast, exhibit only acanthosis nigricans, and sometimes symptomatic postprandial hypoglycemia. Even if noticed, these are much less likely to lead to medical consultation. However, women also have much more severe hyperinsulinemia and dyslipidemia than men. In lipodystrophy, this is almost certainly explained at least in part by the larger amount of white adipose tissue as a proportion of total body mass in healthy women.
VI Biochemical Subphenotyping of Severe IR
Growing evidence suggests that some syndromes of severe IR exhibit different patterns of “IR” among insulin-responsive tissues and pathways. Thus, the generalized IR of insulin receptor (INSR) defects is associated with a normal lipid profile and relative lack of fatty liver (39, 40), suggesting that some insulin signaling is needed to drive hepatic fat synthesis and secretion. This is quite unlike the situation in patients with lipodystrophy or with defects in the insulin signal transducer AKT2, all of whom show severe dyslipidemia and fatty liver (39).
The mechanisms linking hyperinsulinemia in prevalent forms of IR and fatty liver/dyslipidemia are of particular importance, given the enormous associated prevalence of atherosclerosis, and are the subject of intense investigation. Full consideration of progress in the field is beyond the scope of this article; however, recent cell-based studies have strongly implicated activation of the mammalian target of rapamycin complex 1 (mTORC1), hitherto widely perceived to be predominantly a mediator of insulin's actions on cell growth, in driving hepatic de novo lipogenesis in response to insulin (58–60). Such up-regulation of hepatic lipogenesis has been suggested to be a significant contributor to fatty liver and atherogenic dyslipidemia in humans (61, 62), whereas aberrant activation of mTORC1 is well documented in IR (63), potentially explaining why lipogenesis appears to be increased in these states. However, many critical questions remain to be answered, including whether enhanced de novo lipogenesis in IR may truly be explained as a hepatocyte autonomous phenomenon related to resistance to only some of insulin's cellular effects, or whether it may instead reflect parallel hyperactivation of mTORC1 by increased delivery to hepatocytes of, for example, branched chain amino acids, which have been reported to be elevated in many forms of IR (59, 64, 65).
INSR defects may also be discriminated from other forms of IR by unexpectedly high adiponectin (66–68), SHBG (69), and IGF binding protein-1 (IGFBP1) (70) levels, providing further evidence that in prevalent forms of IR (71–73), in lipodystrophies (15, 67), and in nonreceptoropathy severe IR (70, 74), where the levels of these proteins are usually reduced, hyperinsulinemia is able to exert effects through intact elements of the cellular insulin signal transduction network to suppress gene expression in both adipose tissue and liver (62, 75). Parenthetically, because adiponectin is nearly exclusively expressed in adipose tissue and because circulating SHBG and IGFBP1 are products of hepatic expression, determination of these markers in states of IR may also allow assessment to be made of the insulinization of distinct insulin target tissues in future, enhancing clinical ability to discern any organ-selective forms of severe IR.
This concept of IR subphenotypes has been exploited diagnostically by using it to subclassify IR and target genetic screening, greatly enhancing efficiency of molecular diagnosis of INSR defects (68, 70). Thus, in the context of severe IR, we have found that adiponectin levels above 7 mg/liter have a 97 positive predictive value for insulin receptoropathy (70), although the precise cutoff is assay-specific. SHBG and IGFBP1 levels have lesser, although still significant, utility.
VII Monogenic IR Classification/Nomenclature
The nomenclature in the field of severe IR dates from the 1970s. Then, in seminal publications, Kahn and colleagues (9, 16) designated severe IR in nonobese patients as “type A” or “type B,” the latter discriminated by the presence of anti-insulin receptor antibodies. In a series of independent publications around the same time, the term “HAIR-AN” came to be used commonly (76). However, “HAIR-AN” (hyperandrogenism, insulin resistance, and acanthosis nigricans) is an entirely generic description of severe IR in women. If it has any utility, it is where it is used to discriminate women with severe IR who also have a body mass index above 30 kg/m2, who are much less likely to harbor pathogenic single gene defects than their nonobese counterparts. However, the imprecise and overlapping usage of these different diagnostic terms and increasing understanding of subgroups of severe IR suggest that a reclassification of syndromes of severe IR may be timely. Based on the above observations, such a classification is suggested in Table 1.
Proposed new classification for syndromes of severe IR
| Discriminating features | |
|---|---|
| I. Primary insulin-signaling defects | |
| A. Generalizeda [INSR mutations (40) or anti-INSR antibodies (12)] | Extreme hyperinsulinemia but normal lipid profile (39, 40), preserved or elevated adiponectin, SHBG, and IGFBP1 (70) |
| B. Partialb [AKT2 (19), AS160 (91, 169), others to be defined] | Likely to depend upon precise signaling defect |
| II. Secondary to adipose tissue abnormalitiesc | |
| A. Severe obesity [e.g., MC4R,(170),POMC (171), LEP (126), LEPR (172), SH2B1 (173)] | Early onset severe, hyperphagic obesit Tall stature (MC4R) Hypogonadotropic hypogonadism (LEP) Red hair and hypoadrenalism (POMC) Disproportionate IR [SH2B1 (173)] |
| B. Lipodystrophy (generalized or partial, Table 2) | Congenitally absent adipose tissue, or regional deficiency of adipose tissue Usually severe dyslipidemia, fatty liver Low adiponectin and leptin levels |
| Discriminating features | |
|---|---|
| I. Primary insulin-signaling defects | |
| A. Generalizeda [INSR mutations (40) or anti-INSR antibodies (12)] | Extreme hyperinsulinemia but normal lipid profile (39, 40), preserved or elevated adiponectin, SHBG, and IGFBP1 (70) |
| B. Partialb [AKT2 (19), AS160 (91, 169), others to be defined] | Likely to depend upon precise signaling defect |
| II. Secondary to adipose tissue abnormalitiesc | |
| A. Severe obesity [e.g., MC4R,(170),POMC (171), LEP (126), LEPR (172), SH2B1 (173)] | Early onset severe, hyperphagic obesit Tall stature (MC4R) Hypogonadotropic hypogonadism (LEP) Red hair and hypoadrenalism (POMC) Disproportionate IR [SH2B1 (173)] |
| B. Lipodystrophy (generalized or partial, Table 2) | Congenitally absent adipose tissue, or regional deficiency of adipose tissue Usually severe dyslipidemia, fatty liver Low adiponectin and leptin levels |
An alternative term for this cluster of disorders is “insulin receptoropathies.”
Affecting only some intracellular arms of the insulin signaling pathway, or variable among tissues.
In addition to frank obesity or lipodystrophy, there is a less well-defined group of disorders having clinical and biochemical evidence of “adipose tissue failure” and severe dyslipidemia despite grossly normal whole body adipose tissue mass.
Proposed new classification for syndromes of severe IR
| Discriminating features | |
|---|---|
| I. Primary insulin-signaling defects | |
| A. Generalizeda [INSR mutations (40) or anti-INSR antibodies (12)] | Extreme hyperinsulinemia but normal lipid profile (39, 40), preserved or elevated adiponectin, SHBG, and IGFBP1 (70) |
| B. Partialb [AKT2 (19), AS160 (91, 169), others to be defined] | Likely to depend upon precise signaling defect |
| II. Secondary to adipose tissue abnormalitiesc | |
| A. Severe obesity [e.g., MC4R,(170),POMC (171), LEP (126), LEPR (172), SH2B1 (173)] | Early onset severe, hyperphagic obesit Tall stature (MC4R) Hypogonadotropic hypogonadism (LEP) Red hair and hypoadrenalism (POMC) Disproportionate IR [SH2B1 (173)] |
| B. Lipodystrophy (generalized or partial, Table 2) | Congenitally absent adipose tissue, or regional deficiency of adipose tissue Usually severe dyslipidemia, fatty liver Low adiponectin and leptin levels |
| Discriminating features | |
|---|---|
| I. Primary insulin-signaling defects | |
| A. Generalizeda [INSR mutations (40) or anti-INSR antibodies (12)] | Extreme hyperinsulinemia but normal lipid profile (39, 40), preserved or elevated adiponectin, SHBG, and IGFBP1 (70) |
| B. Partialb [AKT2 (19), AS160 (91, 169), others to be defined] | Likely to depend upon precise signaling defect |
| II. Secondary to adipose tissue abnormalitiesc | |
| A. Severe obesity [e.g., MC4R,(170),POMC (171), LEP (126), LEPR (172), SH2B1 (173)] | Early onset severe, hyperphagic obesit Tall stature (MC4R) Hypogonadotropic hypogonadism (LEP) Red hair and hypoadrenalism (POMC) Disproportionate IR [SH2B1 (173)] |
| B. Lipodystrophy (generalized or partial, Table 2) | Congenitally absent adipose tissue, or regional deficiency of adipose tissue Usually severe dyslipidemia, fatty liver Low adiponectin and leptin levels |
An alternative term for this cluster of disorders is “insulin receptoropathies.”
Affecting only some intracellular arms of the insulin signaling pathway, or variable among tissues.
In addition to frank obesity or lipodystrophy, there is a less well-defined group of disorders having clinical and biochemical evidence of “adipose tissue failure” and severe dyslipidemia despite grossly normal whole body adipose tissue mass.
The key subdivision in this proposed mechanism-based classification is between those disorders in which there is a primary defect in canonical insulin signal transduction and those in which severe IR is a consequence of adipose tissue abnormalities, or “adipose failure.” Primary IR is then subdivided into “generalized” IR, in which there is a defect at the level of the insulin receptor, and biochemically distinguishable “partial” IR, in which there is a signaling defect that is limited either to only some parts of the postreceptor signal transduction pathway or to some tissues. Few examples of this group currently exist, but it is to be anticipated that increased recognition of this group, allied to modern sequencing technologies, will lead to further examples and refinement of this subgroup.
Adipose failure may also be subdivided into a group with manifest lipodystrophy, in which there is a deficiency in generating adipose tissue, leading to severe IR despite low or normal adipose tissue mass, and a group in which the dominant defect is unrestrained accumulation of adipose tissue, most commonly due to hyperphagia, such that even a relatively normal capacity safely to accrue triglyceride in adipose tissue is overcome.
Severe IR is also seen in a group of “complex disorders” (see Table 3), in association with other defects; however, because the IR in these conditions is subjected to mechanistic study with the above framework in mind, we anticipate that they may be accommodated within one of the two main groups.
VIII The INSR Spectrum
The first defects in the INSR were reported in 1988 (77, 78), shortly after cloning of the human gene (79), and more than 100 allelic variants have now been described. These genetic insulin receptoropathies form a continuum of clinical severity but are best divided into two groups (Fig. 4). The first consists of rare and severe autosomal recessive (AR) disorders presenting in the first decade of life and usually classified, arbitrarily, as Donohue syndrome (DS; formerly “leprechaunism”) or Rabson Mendenhall syndrome (RMS), based on the original clinical descriptions (80, 81). These syndromes have been well described (40, 82). As well as fasting hypoglycemia, postprandial hyperglycemia, and extreme hyperinsulinemia, their dominant features are markedly retarded linear growth, impaired muscle and adipose tissue development, and overgrowth or precocious development of sex hormone-dependent tissues such as genitalia and nipples, and of other tissues including hair, skin, and viscera (Fig. 4). When β-cells decompensate, hyperglycemia may become refractory to treatment. In DS, death usually occurs during intercurrent infection in infancy, whereas in RMS, advanced microvascular complications or diabetic ketoacidosis are the commonest modes of death, usually in the second or third decade (40).
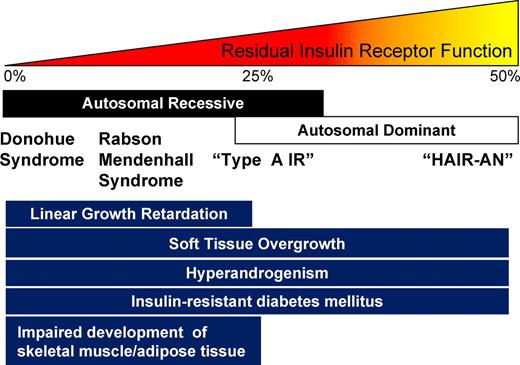
Clinical spectrum of insulin receptoropathies.
Some aspects of the DS phenotype remain to be fully explained. In particular, it remains to be determined definitively why affected infants are resistant to ketoacidosis at least in the first year of life, even in those with no functional insulin receptor, although suggestions include continued action of extremely elevated insulin on persisting hepatic IGF-I receptors in the immature liver or deficiency of GH secretion or action (83).
More commonly, INSR defects present peripubertally as oligomenorrhea and hyperandrogenism with acanthosis nigricans (40). At presentation, hyperglycemia has often yet to develop. In the prediabetic phase, males exhibit only acanthosis nigricans and sometimes hypoglycemia, and they often remain undiagnosed even after the development of symptomatic diabetes, which may not occur until the fourth decade or beyond.
In some patients with DS or RMS, loss of INSR expression from one allele has been identified and has been inferred to be due to a mutation in a regulatory sequence such as the promoter (84). However, four patients with severe IR and extremely low expression of both alleles of the INSR gene have also been described harboring either a heterozygous deletion or mutation of the HMGA1 gene, but no mutations in the INSR gene (85). HMGA1 is an architectural transcription factor that binds to key sites in the promoter of the INSR gene to facilitate its transcription. Based on the single report to date, these patients appeared to share many characteristics of patients with INSR defects, consistent with the notion that a key function for HMGA1 is the stimulation of INSR expression.
IX Downstream Insulin Signaling Defects
Rapid progress in elucidating the key components of the insulin signaling pathway in the early 1990s raised hope that defects may be found in the genes encoding these signaling elements in patients with severe IR but without INSR mutations. However, few such sequence variants have been reported in severe IR, and in most cases the resulting signaling defects in vitro have been subtle at best (86–90).
One exception was a single family in which three members carried a nonfunctional, heterozygous mutation in AKT2, encoding a critical serine/threonine kinase downstream from the INSR in the signal transduction pathway (19). Clinical features seen in affected family members included acanthosis nigricans, ovarian hyperandrogenism, diabetes mellitus presaged by several years of postprandial hypoglycemia, metabolic dyslipidemia, and fatty liver (19). The female proband also exhibited partial lipodystrophy, highlighting the role of insulin in adipogenesis and the need for awareness that primary defects in the insulin signaling cascade may impair adipose tissue generation in vivo. However, although a generalized deficiency in adipose tissue development is seen in severe insulin receptoropathies, the metabolic characteristics of this differ dramatically from the “fat failure” phenotype of primary generalized lipodystrophy, and they may more appropriately be regarded as states of nonlipidated rather than absent adipose tissue.
More recently, a heterozygous nonsense mutation in AS160, a small GTPase-activating protein that forms a key link between insulin signaling and glucose uptake by the GLUT4 transporter, has been described in a family in whom affected members had acanthosis nigricans and disproportionate hyperinsulinemia after a glucose challenge (91). Surprisingly, no other mutations have been found to date in other, more distal signaling components involved in GLUT4 transporter translocation to the cell membrane in response to insulin.
X Disorders of Adipose Tissue Development/Function (Lipodystrophies)
Far more success has been had in identifying primary defects in adipose tissue that lead to severe IR as a secondary consequence. This probably reflects the fact that lipodystrophy, in contrast to many other forms of severe IR, is relatively easily recognized clinically, facilitating identification of extended families with multiple affected members for genetic studies (92–96). The possibility of lipodystrophy should be carefully considered in all severely insulin-resistant patients with dyslipidemia and/or nonalcoholic fatty liver disease.
Lipodystrophy is a heterogeneous disorder characterized by pathological adipose tissue deficiency. The lack of fat may be partial or generalized and inherited or acquired in origin. The molecular pathogenesis and clinical features of lipodystrophy have recently been reviewed (15, 97–101), are not covered in detail here, but are summarized in Table 2. Within the past 2 yr, three novel subtypes of congenital lipodystrophy were identified by a candidate gene approach. Biallelic nonsense mutations in CAV1 (102) and PTRF (103–106) were identified in patients with generalized lipodystrophy and in CIDEC in a patient with partial lipodystrophy (107). Caveolins are essential for the formation of caveolae, which are abundant in adipocytes and appear to play a role in fatty acid uptake, insulin receptor signaling (108), and lipid droplet formation (109). PTRF (polymerase 1 and transcript release factor) stabilizes caveolins 1–3 and is required for the formation of caveolae (110, 111). CAV3 mutations are known to cause muscular dystrophy (112), which probably explains why PTRF mutations were also associated with a muscular dystrophy phenotype.
Classification and clinical features of lipodystrophies
| Inheritance | Major clinical features | Ref. | |
|---|---|---|---|
| CGL | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 15 | |
| AGPAT2 | AR | Adiponectin levels are particularly low in this form of CGL, whereas they are slightly higher (although still lower than the reference range) in BSCL2-associated CGL | 67,174 |
| BSCL2a | AR | See note above | 174 |
| CAV1a | AR (single case) | Short stature | 102 |
| PTRFa | AR | Muscular dystrophy, modest metabolic disturbance (?) | 103–106 |
| Familial partial lipodystrophy | Common features: IR, T2DM, dyslipidemia, fatty liver, PCOS | ||
| LMNAa | AD | Preserved/excess facial and neck fat | 15, 175 |
| PPARGa | AD | Preserved abdominal fat, hypertension (?) | 18,176,177–179 |
| ZMPSTE24a | AR | Mandibuloacral dysplasia | |
| AKT2a | AD (single family) | 19 | |
| CIDECa | AR (single case) | Preserved facial and neck fat, multiloculated lipid droplets | 107 |
| Acquired generalized lipodystrophy | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 14, 15 | |
| Associated with other autoimmune diseases; commonly also associated with low C4 complement levels | N/A | May be associated with juvenile dermatomyositis, SLE, autoimmune hemolytic anemia, autoimmune hepatitis. The low C4 complement subgroup is particularly associated with autoimmune hepatitis and autoimmune hemolytic anemia. | 11, 13, 14 |
| Acquired partial lipodystrophy | 14 | ||
| HIV-associated lipodystrophy | N/A | Not typically associated with “severe” IR but is associated with IR, dyslipidemia, fatty liver | 180 |
| C3 nephritic factor associated | N/A | Cephalocaudal pattern of fat loss, low C3 complement levels, MPGN, not usually insulin resistant, although may become so if overweight | 14 |
| Inheritance | Major clinical features | Ref. | |
|---|---|---|---|
| CGL | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 15 | |
| AGPAT2 | AR | Adiponectin levels are particularly low in this form of CGL, whereas they are slightly higher (although still lower than the reference range) in BSCL2-associated CGL | 67,174 |
| BSCL2a | AR | See note above | 174 |
| CAV1a | AR (single case) | Short stature | 102 |
| PTRFa | AR | Muscular dystrophy, modest metabolic disturbance (?) | 103–106 |
| Familial partial lipodystrophy | Common features: IR, T2DM, dyslipidemia, fatty liver, PCOS | ||
| LMNAa | AD | Preserved/excess facial and neck fat | 15, 175 |
| PPARGa | AD | Preserved abdominal fat, hypertension (?) | 18,176,177–179 |
| ZMPSTE24a | AR | Mandibuloacral dysplasia | |
| AKT2a | AD (single family) | 19 | |
| CIDECa | AR (single case) | Preserved facial and neck fat, multiloculated lipid droplets | 107 |
| Acquired generalized lipodystrophy | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 14, 15 | |
| Associated with other autoimmune diseases; commonly also associated with low C4 complement levels | N/A | May be associated with juvenile dermatomyositis, SLE, autoimmune hemolytic anemia, autoimmune hepatitis. The low C4 complement subgroup is particularly associated with autoimmune hepatitis and autoimmune hemolytic anemia. | 11, 13, 14 |
| Acquired partial lipodystrophy | 14 | ||
| HIV-associated lipodystrophy | N/A | Not typically associated with “severe” IR but is associated with IR, dyslipidemia, fatty liver | 180 |
| C3 nephritic factor associated | N/A | Cephalocaudal pattern of fat loss, low C3 complement levels, MPGN, not usually insulin resistant, although may become so if overweight | 14 |
AD, Autosomal dominant; CGL, congenital generalized lipodystrophy; C3/4, complement factor 3/4; T2DM, type 2 diabetes mellitus; PCOS, polycystic ovary syndrome; MPGN, mesangioproliferative glomerulonephritis; N/A, not applicable; SLE, systemic lupus erythematosus.
Genetic subtype.
Classification and clinical features of lipodystrophies
| Inheritance | Major clinical features | Ref. | |
|---|---|---|---|
| CGL | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 15 | |
| AGPAT2 | AR | Adiponectin levels are particularly low in this form of CGL, whereas they are slightly higher (although still lower than the reference range) in BSCL2-associated CGL | 67,174 |
| BSCL2a | AR | See note above | 174 |
| CAV1a | AR (single case) | Short stature | 102 |
| PTRFa | AR | Muscular dystrophy, modest metabolic disturbance (?) | 103–106 |
| Familial partial lipodystrophy | Common features: IR, T2DM, dyslipidemia, fatty liver, PCOS | ||
| LMNAa | AD | Preserved/excess facial and neck fat | 15, 175 |
| PPARGa | AD | Preserved abdominal fat, hypertension (?) | 18,176,177–179 |
| ZMPSTE24a | AR | Mandibuloacral dysplasia | |
| AKT2a | AD (single family) | 19 | |
| CIDECa | AR (single case) | Preserved facial and neck fat, multiloculated lipid droplets | 107 |
| Acquired generalized lipodystrophy | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 14, 15 | |
| Associated with other autoimmune diseases; commonly also associated with low C4 complement levels | N/A | May be associated with juvenile dermatomyositis, SLE, autoimmune hemolytic anemia, autoimmune hepatitis. The low C4 complement subgroup is particularly associated with autoimmune hepatitis and autoimmune hemolytic anemia. | 11, 13, 14 |
| Acquired partial lipodystrophy | 14 | ||
| HIV-associated lipodystrophy | N/A | Not typically associated with “severe” IR but is associated with IR, dyslipidemia, fatty liver | 180 |
| C3 nephritic factor associated | N/A | Cephalocaudal pattern of fat loss, low C3 complement levels, MPGN, not usually insulin resistant, although may become so if overweight | 14 |
| Inheritance | Major clinical features | Ref. | |
|---|---|---|---|
| CGL | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 15 | |
| AGPAT2 | AR | Adiponectin levels are particularly low in this form of CGL, whereas they are slightly higher (although still lower than the reference range) in BSCL2-associated CGL | 67,174 |
| BSCL2a | AR | See note above | 174 |
| CAV1a | AR (single case) | Short stature | 102 |
| PTRFa | AR | Muscular dystrophy, modest metabolic disturbance (?) | 103–106 |
| Familial partial lipodystrophy | Common features: IR, T2DM, dyslipidemia, fatty liver, PCOS | ||
| LMNAa | AD | Preserved/excess facial and neck fat | 15, 175 |
| PPARGa | AD | Preserved abdominal fat, hypertension (?) | 18,176,177–179 |
| ZMPSTE24a | AR | Mandibuloacral dysplasia | |
| AKT2a | AD (single family) | 19 | |
| CIDECa | AR (single case) | Preserved facial and neck fat, multiloculated lipid droplets | 107 |
| Acquired generalized lipodystrophy | Common features: severe IR, T2DM, severe dyslipidemia, fatty liver, pseudoacromegaly, PCOS | 14, 15 | |
| Associated with other autoimmune diseases; commonly also associated with low C4 complement levels | N/A | May be associated with juvenile dermatomyositis, SLE, autoimmune hemolytic anemia, autoimmune hepatitis. The low C4 complement subgroup is particularly associated with autoimmune hepatitis and autoimmune hemolytic anemia. | 11, 13, 14 |
| Acquired partial lipodystrophy | 14 | ||
| HIV-associated lipodystrophy | N/A | Not typically associated with “severe” IR but is associated with IR, dyslipidemia, fatty liver | 180 |
| C3 nephritic factor associated | N/A | Cephalocaudal pattern of fat loss, low C3 complement levels, MPGN, not usually insulin resistant, although may become so if overweight | 14 |
AD, Autosomal dominant; CGL, congenital generalized lipodystrophy; C3/4, complement factor 3/4; T2DM, type 2 diabetes mellitus; PCOS, polycystic ovary syndrome; MPGN, mesangioproliferative glomerulonephritis; N/A, not applicable; SLE, systemic lupus erythematosus.
Genetic subtype.
One female patient with partial lipodystrophy (affecting limb, femorogluteal, and sc abdominal fat), white adipocytes with multiloculated lipid droplets, and insulin-resistant diabetes was found to be homozygous for a premature truncation mutation in the lipid droplet protein, CIDEC (cell death-inducing DNA fragmentation factor A-like effector family-C) (107). Cidec knockdown cells manifest multiloculated lipid droplets with increased mitochondria (113), and in mice, Cidec deficiency also reduces fat mass and induces the formation of white adipocytes with multilocular lipid droplets (114, 115). However, in contrast to the human phenotype associated with a homozygous loss of function mutation in CIDEC, Cidec null mice are protected against diet-induced obesity and IR (114–116). BSCL2, an enigmatic gene of unknown function in which homozygous mutations were the first identified cause of congenital lipodystrophy (95), has also recently been implicated in lipid droplet biogenesis (117–119) and adipocyte differentiation (120).
XI Digenic IR
In 2002 (121), we described a family in which five severely insulin-resistant subjects and no unaffected relatives were doubly heterozygous for frameshift/premature stop mutations in two unlinked genes, namely PPARG, a key regulator of adipocyte biology, and PPP1R3A, a muscle-specific protein involved in regulating glycogen turnover (122–125). This report was of particular interest because the observation that genetic defects in molecules primarily involved in either lipid or carbohydrate metabolism can combine to result in an extreme phenotype of IR provides a model for the type of metabolic interaction that may underlie common forms of IR/type 2 diabetes.
XII Complex Syndromes
Several genetic syndromes feature severe IR as part of a wider constellation of abnormality (Table 3). In many of these, the IR is related to severe obesity. However, this group may roughly be divided into those conditions in which IR does not seem to be disproportionate to the degree of excess adiposity, such as genetically severe hyperphagia due to congenital leptin deficiency (126), and those in which IR appears unusually severe, suggesting a role for the defective genes concerned in systemic insulin sensitivity as well as in hypothalamic appetite control. However, in most cases this issue has not been rigorously studied. One well-established example is Alström syndrome, which is due to genetic defects in the large centrosomal ALMS1 protein, where predominantly centripetal adiposity is associated with severely disproportionate IR and dyslipidemia (127).
Selected complex genetic disorders associated with severe IR
| Syndrome | Gene(s) | Adipose tissue phenotype | IR disproportionate to adiposity? | Cellular component or function affected |
|---|---|---|---|---|
| Alstöm | ALMS1 | Centripetal obesity | Yes (127) | Centrosome/ basal body |
| MOPDII | PCNT | Centripetal fat distribution | Yes (128) | Centrosome/basal body |
| Bardet Biedl | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, BBS8, BBS9, BBS10, BBS11, BBS12, MKS1, CEP290 | Obesity | Unclear (129) | Centrosome/basal body (130) |
| Bloom | RECQ2 | Lipodystrophy | Yes (133) | DNA repair |
| Werner | RECQL2 | Lipodystrophy | Yes (131–132) | DNA repair |
| LMNA | ||||
| Mandibuloacral dysplasia | LMNA | Lipodystrophy | Yes (97, 134) | Involved in formation of nuclear lamina |
| ZMPSTE24 | ||||
| Myotonic dystrophy | DMPK | None | Yes (181) | Transcriptional/splicing regulation on chromosome 19, including the INSR (182) |
| Syndrome | Gene(s) | Adipose tissue phenotype | IR disproportionate to adiposity? | Cellular component or function affected |
|---|---|---|---|---|
| Alstöm | ALMS1 | Centripetal obesity | Yes (127) | Centrosome/ basal body |
| MOPDII | PCNT | Centripetal fat distribution | Yes (128) | Centrosome/basal body |
| Bardet Biedl | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, BBS8, BBS9, BBS10, BBS11, BBS12, MKS1, CEP290 | Obesity | Unclear (129) | Centrosome/basal body (130) |
| Bloom | RECQ2 | Lipodystrophy | Yes (133) | DNA repair |
| Werner | RECQL2 | Lipodystrophy | Yes (131–132) | DNA repair |
| LMNA | ||||
| Mandibuloacral dysplasia | LMNA | Lipodystrophy | Yes (97, 134) | Involved in formation of nuclear lamina |
| ZMPSTE24 | ||||
| Myotonic dystrophy | DMPK | None | Yes (181) | Transcriptional/splicing regulation on chromosome 19, including the INSR (182) |
MOPDII, Osteodysplastic primordial dwarfism of Majewski type II.
Selected complex genetic disorders associated with severe IR
| Syndrome | Gene(s) | Adipose tissue phenotype | IR disproportionate to adiposity? | Cellular component or function affected |
|---|---|---|---|---|
| Alstöm | ALMS1 | Centripetal obesity | Yes (127) | Centrosome/ basal body |
| MOPDII | PCNT | Centripetal fat distribution | Yes (128) | Centrosome/basal body |
| Bardet Biedl | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, BBS8, BBS9, BBS10, BBS11, BBS12, MKS1, CEP290 | Obesity | Unclear (129) | Centrosome/basal body (130) |
| Bloom | RECQ2 | Lipodystrophy | Yes (133) | DNA repair |
| Werner | RECQL2 | Lipodystrophy | Yes (131–132) | DNA repair |
| LMNA | ||||
| Mandibuloacral dysplasia | LMNA | Lipodystrophy | Yes (97, 134) | Involved in formation of nuclear lamina |
| ZMPSTE24 | ||||
| Myotonic dystrophy | DMPK | None | Yes (181) | Transcriptional/splicing regulation on chromosome 19, including the INSR (182) |
| Syndrome | Gene(s) | Adipose tissue phenotype | IR disproportionate to adiposity? | Cellular component or function affected |
|---|---|---|---|---|
| Alstöm | ALMS1 | Centripetal obesity | Yes (127) | Centrosome/ basal body |
| MOPDII | PCNT | Centripetal fat distribution | Yes (128) | Centrosome/basal body |
| Bardet Biedl | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, BBS8, BBS9, BBS10, BBS11, BBS12, MKS1, CEP290 | Obesity | Unclear (129) | Centrosome/basal body (130) |
| Bloom | RECQ2 | Lipodystrophy | Yes (133) | DNA repair |
| Werner | RECQL2 | Lipodystrophy | Yes (131–132) | DNA repair |
| LMNA | ||||
| Mandibuloacral dysplasia | LMNA | Lipodystrophy | Yes (97, 134) | Involved in formation of nuclear lamina |
| ZMPSTE24 | ||||
| Myotonic dystrophy | DMPK | None | Yes (181) | Transcriptional/splicing regulation on chromosome 19, including the INSR (182) |
MOPDII, Osteodysplastic primordial dwarfism of Majewski type II.
Although the molecular pathogenesis of severe IR in Alström syndrome is not clear, it is notable that defects in the pericentrosomal protein pericentrin, causing osteodysplastic primordial dwarfism of Majewski type 2, are also associated with highly penetrant severe IR (128), which may also be true in many cases of Bardet Biedl syndrome (129), caused by defects in a variety of proteins involved in basal body/centrosomal function (130). Collectively, these observations hint at an important role for the centrosome or basal body, or the cellular functions they subserve, in maintaining metabolic homeostasis.
Another notable group of disorders that feature disproportionate and often severe IR are associated with DNA repair defects and/or progeria, including Werner syndrome (131, 132), Bloom syndrome (133), and mandibuloacral dysplasia (97, 134). In further DNA repair defects such as ataxia telangiectasia, severe IR has been reported in several molecularly defined cases (135, 136). However, the precise mechanism of severe IR in these settings has yet to be established.
XIII Therapy
The rarity and underdiagnosis of severe IR means that almost all therapeutic decisions are based on rational targeting of underlying defects and anecdotal evidence rather than randomized controlled trials. Therapy aims to reduce hyperglycemia, to ameliorate dyslipidemia, and to lessen the sometimes debilitating reproductive and cosmetic consequences of hyperinsulinemia. This is achieved first through mitigation of the underlying signaling defect, through minimizing secretory demands on the pancreatic β-cells, and through optimal delivery of exogenous insulin when required. In the context of adipose tissue absence or dysfunction, offloading adipose tissue by reducing lipid delivery to it is also critical, and, finally, countering hyperandrogenism, ovulatory dysfunction, and acanthosis nigricans by measures targeted at the relevant end organs is also often of value. The treatment of such secondary manifestations of the IR state has been reviewed elsewhere (137, 138).
A Dietary and lifestyle modification
In severe IR, whether or not a single gene defect is identified, weight gain inevitably exacerbates metabolic derangement and either worsens hyperglycemia in patients with overt diabetes or increases β-cell stress, so restricting energy intake and maximizing aerobic exercise are essential elements of management. This is particularly important in lipodystrophy where the apparent leanness of patients frequently results in a failure of caregivers to place sufficient emphasis on dietary modification. Indeed, failure to restrict energy intake in patients with lipodystrophy makes it almost impossible to obtain good glycemic and lipidemic control.
B Insulin sensitization and replacement
Insulin-sensitizing agents also play a key role in management. Metformin is often effective, and in some cases thiazolidinediones also exert markedly beneficial effects, but no comparative studies exist to guide the choice of therapy in different subgroups of severe IR. When β-cell decompensation occurs in severe IR to produce diabetes, this is only relative to the very high insulin requirements, and plasma insulin levels remain extremely elevated. This means that insulin secretagogues such as sulfonylureas often produce little benefit. When insulin is required, this may need to be used in concentrated form to achieve metabolic control (139), and limited evidence suggests that delivery by sc infusion may be efficacious (140). Use of recombinant human IGF-I has been reported mostly in the setting of severe insulin receptoropathies and appears to improve glycemia and perhaps survival in some infantile cases (141–151). It may exert activity by acting as an insulin mimetic, as a trophic factor for pancreatic β-cells, or by enhancing insulin sensitivity through postreceptor cross talk between insulin and IGF-I signaling pathways. However, its dominant mode of action, optimal dosing, and clinical indications remain unclear.
C Adipose tissue offloading
Minimizing caloric intake and hence strain on adipose storage capacity is particularly difficult in lipodystrophy because either absolute or relative leptin deficiency, in generalized and partial lipodystrophy, respectively, leads to hyperphagia (152). Recombinant leptin therapy substantially reduces food intake in this setting and dramatically improves dyslipidemia, hepatic steatosis, and glycemic control (153–157). The response to leptin and dose titration is largely based on clinical criteria because many patients develop antibodies that interfere with leptin assays (158) but do not, on the whole, appear to reduce its long-term efficacy (159). Leptin has been used in all prevalent forms of generalized and familial partial lipodystrophy, with particularly dramatic results in the former (157). Initial reports also suggest that it may be useful in at least some cases of HIV-associated partial lipodystrophy (160), whereas a single study has also reported benefits in RMS (161). In principle, increasing oxidative catabolism of excess calories may achieve the same benefits as limiting intake, as illustrated by the dramatically beneficial effect of suppressive doses of T4 in a patient with an INSR defect who also had a papillary thyroid carcinoma (162), but this strategy has yet to be developed safely for more widespread application.
Given the importance of restricting energy intake, particularly in patients with lipodystrophy, other weight loss therapies used in obese diabetic patients, including glucagon-like peptide-1 agonists and appetite suppressants, have the potential to produce clinical benefits (163), and scattered reports have even suggested that bariatric surgery can be helpful in severe cases (164).
A complementary strategy to reducing the energy input into adipose tissue is to increase its storage capacity by using insulin-sensitizing thiazolidinedione peroxisome proliferator-activated receptor γ agonists. These were an obvious choice, particularly in lipodystrophy where it was hoped that they might restore fat mass. However, thiazolidinediones are not helpful in generalized lipodystrophy and exacerbate hepatic steatosis in animal models (165), and reports of their use in partial lipodystrophy conflict (166–168). Our own experience is similar to that of Simha et al. (167), who noted that fat tended to accumulate in residual adipose depots, with modest metabolic benefits.
XIV Summary
Genetic syndromes of severe IR, with or without lipodystrophy, are underrecognized conditions that exact an immense toll of early morbidity and mortality for those affected. Considerable progress over the past 20 yr in identifying the molecular basis of these disorders now presents the opportunity for trials or therapy targeted at specific subgroups of these patients. It is anticipated that this will not only improve clinical outcomes for these rare patients but will also give insights into the pathophysiology and therapy of more prevalent forms of IR.
Acknowledgments
This work was supported by research grants from the Wellcome Trust (Intermediate Clinical Fellowship 080952/Z/06/Z, to R.K.S.; Programme Grant 078986/Z/06/Z, to S.O.), GlaxoSmithKline (to D.B.S.), the UK National Institute for Health Research Cambridge Biomedical Research Centre, and the UK Medical Research Council Centre for Obesity and Related Metabolic Disease.
Disclosure Summary: R.K.S., D.B.S., E.K.C., and P.G. have nothing to disclose. S.O. acts as a consultant in drug discovery for GlaxoSmithKline, Pfizer, and OSI Pharmaceuticals, Inc.
Abbreviations
- AR
Autosomal recessive
- CIDEC
cell death-inducing DNA fragmentation factor A-like effector family-C
- DS
Donohue syndrome
- IGFBP1
IGF binding protein-1
- INSR
insulin receptor
- IR
insulin resistance
- mTORC1
mammalian target of rapamycin complex 1
- PTRF
polymerase 1 and transcript release factor
- RMS
Rabson Mendenhall syndrome
References
Author notes
R.K.S. and D.B.S. contributed equally to this work

{kind=link}
{kind=link}
{kind=link}
{kind=link}